2. From Pairs to Crystals: Many-Body Effects
Moving from isolated pairs to clusters and crystals reveals fundamental challenges in computational chemistry. The total energy is not simply the sum of pairwise interactions. This section explores how to calculate lattice energies using the occ elat command and estimate energies with fast semi-empirical methods like xTB.
The Many-Body Problem
Why Pairs Don't Tell the Whole Story
Consider three water molecules A, B, and C. The total interaction energy is:
where is the non-additive three-body term. This arises from:
- Polarization effects: A polarizes B, which affects how B interacts with C
- Charge transfer: Electron density redistributes across all three molecules
- Exchange repulsion: Overlapping electron clouds of all three molecules
Energy Hierarchy in Clusters
For a cluster of N molecules, the many-body expansion is:
Where:
- = pairwise interactions (typically 85-95% of total)
- = three-body corrections (5-15%)
- Higher terms usually < 1%
Physical Origins of Many-Body Effects
The breakdown of pairwise additivity occurs because molecular environments are not independent. When molecule A interacts with molecule B, it changes how A can subsequently interact with molecule C. This leads to several physical effects:
1. Cooperative Effects (Negative 3-body terms)
- Polarization enhancement: A polarizes B, making B a better hydrogen bond partner for C
- Charge transfer cascades: Electrons flow A→B→C, strengthening all interactions
- Common in: π-stacking, metal coordination, extended conjugated systems
2. Anti-cooperative Effects (Positive 3-body terms)
- Polarization saturation: A polarizes B, leaving B less able to interact with C
- Electrostatic screening: Charges reorganize to minimize unfavorable interactions
- Common in: Hydrogen bonding networks, ionic solutions
3. Mixed Effects
- Real systems often show both types depending on geometry and chemical environment
- Water clusters typically show weak anti-cooperativity (~5% of binding)
- Overall magnitude determines whether pairwise models are adequate
The key insight is that many-body effects are not just corrections - they reveal fundamental physics about how molecular interactions depend on chemical environment.
The Crystal Challenge
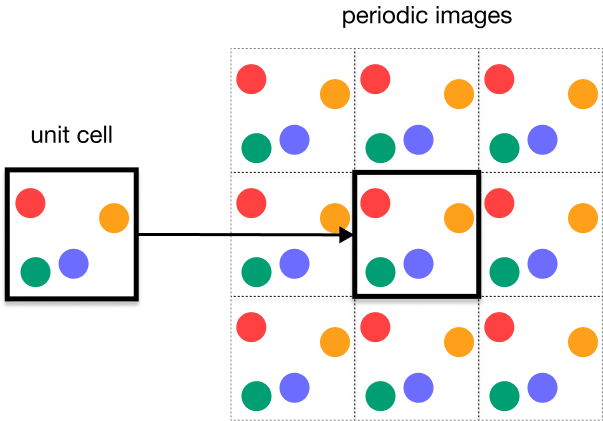
In a crystal, we face additional complexity:
- Infinite interactions: Each molecule interacts with infinite neighbors
- Long-range electrostatics: Decay as , requiring special summation techniques
- Periodic boundary conditions: The unit cell represents infinite repetition
The lattice energy is:
The factor of 1/2 avoids double counting. The sum converges because:
- Electrostatic: Conditionally convergent (depends on summation order)
- Dispersion: Converges as
- Total molecules within sphere of radius R:
- Total dispersion energy: → converges
Computational Exercises
The exercises below demonstrate the concepts outlined above using real calculations. We'll progress from small clusters (where we can calculate everything exactly) to larger systems (where approximations become necessary) to infinite crystals (where pairwise models excel).
Exercise 1: Many-Body Effects in Water Trimers
This exercise quantifies the breakdown of pairwise additivity for a realistic water trimer extracted from ice crystal structure. We'll calculate all the individual components of the many-body expansion to see how significant three-body effects really are.
Many-Body Effects in Water Trimers
Calculate three-body contributions to water trimer interaction energy to understand the limitations of pairwise additivity.
Step 1: Navigate to water trimers directory
Instructions
Navigate to the water trimers directory and examine the molecular structures extracted from ice crystal.
Commands
cd awscc_workshop_2025/02_pairwise_sums/water_trimers/
ls -la *.xyz
echo "---"
echo "Number of atoms in each file:"
wc -l *.xyz | grep -v totalExpected Output
A.xyz B.xyz C.xyz AB.xyz AC.xyz BC.xyz ABC.xyz
---
Number of atoms in each file:
3 A.xyz
3 B.xyz
3 C.xyz
6 AB.xyz
6 AC.xyz
6 BC.xyz
9 ABC.xyzNotes
We have 3 monomers (A, B, C), 3 dimers (AB, AC, BC), and 1 trimer (ABC) from ice structure.
Exercise 2: Convergence of Interaction Energy with Cluster Size
Moving from trimers to larger clusters, we need to understand how interaction energies converge with system size. This is crucial for simulation cutoffs and for understanding when finite cluster calculations approximate bulk behavior. We'll use fast semi-empirical xTB calculations to study clusters with dozens of molecules.
Ice Cluster Convergence Study
Calculate interaction energies for ice clusters with 4 and 8 neighbor shells to understand the range of intermolecular interactions.
Step 1: Navigate to ice clusters directory
Instructions
Navigate to the ice clusters directory and examine the pre-built cluster structures from ice crystal.
Commands
cd ../ice_clusters/
ls -la *.xyz
echo "---"
echo "Number of water molecules in each file:"
for f in *.xyz; do echo "$f: $(head -1 $f) molecules"; doneExpected Output
ice_central_molecule.xyz ice_cluster_4.xyz ice_cluster_8.xyz
ice_neighbors_4.xyz ice_neighbors_8.xyz ice_cluster_12.xyz ice_cluster_16.xyz
---
Number of water molecules in each file:
ice_central_molecule.xyz: 3 molecules
ice_cluster_4.xyz: 15 molecules
ice_cluster_8.xyz: 27 molecules
ice_neighbors_4.xyz: 12 molecules
ice_neighbors_8.xyz: 24 molecules
ice_cluster_12.xyz: 45 molecules
ice_cluster_16.xyz: 65 moleculesNotes
Clusters are built with a central molecule plus neighbor shells. E_interaction = E_cluster - E_central - E_neighbors.
Understanding Energy Units in Crystals
Always specify what your energy is per unit of. This is one of the most common sources of error in computational crystal studies!
Different Ways to Report Crystal Energies
- Per molecule: Energy to remove one molecule from crystal
- Per unit cell: Energy for the entire repeating unit
- Per asymmetric unit: Energy for symmetry-unique part
- Per formula unit: Energy for the simplest stoichiometric unit
- Per atom: Often used in benchmarking studies for fair comparison
Common Mistakes and How to Avoid Them
Example 1: Ice vs. Salt
- Ice: H₂O → 1 molecule per formula unit
- NaCl: Na⁺ + Cl⁻ → 2 ions per formula unit
- Comparing "-800 kJ/mol" for NaCl vs "-63 kJ/mol" for ice is meaningless!
- Must compare: NaCl (-400 kJ/mol per ion) vs H₂O (-63 kJ/mol per molecule)
Example 2: Co-crystals and Solvates
- Paracetamol monohydrate: (C₈H₉NO₂)·(H₂O) → 2 molecules per formula unit
- Pure paracetamol: C₈H₉NO₂ → 1 molecule per formula unit
- To compare stability, normalize per paracetamol molecule, not per formula unit
Example 3: Polymorphs
- Form I: 2 molecules in asymmetric unit → divide by 2 for per-molecule energy
- Form II: 4 molecules in asymmetric unit → divide by 4 for per-molecule energy
- Only then can you compare which polymorph is more stable
For ice example:
- Asymmetric unit energy: -125.6 kJ/mol (contains 2 H₂O)
- Per formula unit (H₂O): -62.8 kJ/mol per molecule
- Experimental sublimation: +54.1 kJ/mol per molecule (opposite sign)
Why benchmarking studies use "per atom" units: Many computational method comparisons report errors in meV/atom rather than per unit cell or per molecule. This allows fair comparison across:
- Small molecules (few atoms) vs. large molecules (many atoms)
- Different crystal packing (1 vs. 8 molecules per unit cell)
- Various stoichiometries (AB vs. A₂B₃ compounds)
For example, a state-of-the-art method like PET-MAD achieves ~20 meV/atom error:
- H₂O (3 atoms): 60 meV = 5.8 kJ/mol of water molecules
- Paracetamol (20 atoms): 400 meV = 38.6 kJ/mol of paracetamol molecules
For the most accurate bespoke dataset, errors can be as low as 3 meV/atom:
- H₂O (3 atoms): 9 meV = 0.9 kJ/mol of water molecules
- Paracetamol (20 atoms): 60 meV = 5.8 kJ/mol of paracetamol molecules
- Both represent the same relative accuracy per atom
Connecting to Experiments: Sublimation Thermodynamics
For molecular crystals, the experimental sublimation enthalpy relates to our calculated lattice energy:
The 2RT term (~5 kJ/mol at 298K) accounts for the difference between constrained vibrations in the crystal and free translation/rotation in the gas phase. For ice:
- Calculated = -62.8 kJ/mol
- Predicted = 62.8 - 4.96 = 57.8 kJ/mol
- Experimental = 54.1 kJ/mol (7% error)
This excellent agreement validates the CE-1p approach for molecular crystals.
Exercise 3: Lattice Energy and Crystal Cohesion
Finally, we tackle crystals using the CE-1P pairwise summation approach. This exercise demonstrates how pairwise models can work surprisingly well for molecular crystals, despite the many-body effects we've just studied. The key insight is that while individual three-body terms matter, their collective effect in crystals may well cancel and is often well-approximated by pairwise models.
Ice Lattice Energy Calculation
Calculate the lattice energy of ice Ih crystal using pairwise CE-1P interactions to understand crystal cohesion and predict sublimation enthalpy.
Step 1: Navigate to ice lattice energy directory
Instructions
Navigate to the ice_elat directory containing the crystal structure file and examine the setup.
Commands
cd ../ice_elat/
ls -la
head -20 ice.cif"Expected Output
ice.cif run_ice_elat.sh example_ice_stdout
data_H2O
_symmetry_space_group_name_H-M P6_3cm
_cell_length_a 7.60356630
_cell_length_b 7.60356630
_cell_length_c 7.14296200
_cell_angle_alpha 90.00000000
_cell_angle_beta 90.00000000
_cell_angle_gamma 120.00000000
_symmetry_Int_Tables_number 185
_chemical_formula_structural H2O
_chemical_formula_sum 'H24 O12'
_cell_volume 357.63798996Notes
Ice Ih has hexagonal symmetry (space group P6_3cm) with a ≈ 7.6 Å and c ≈ 7.14 Å.
Connecting Theory to Practice: What We've Learned
The three exercises above demonstrate key principles of many-body interactions and crystal energetics:
From Exercise 1 (Water Trimers):
- Many-body effects are real but modest (~5%) for hydrogen-bonded systems, trimers, quatermers etc. are still probably not representative of a bulk...
- Individual three-body terms can be calculated exactly for small systems
From Exercise 2 (Cluster Convergence):
- Simulation cutoffs represent accuracy vs. cost tradeoffs
- Different properties (energies vs. forces vs. phase behavior) may require different cutoffs
From Exercise 3 (Crystal Lattice Energies):
- Pairwise models can work remarkably well (~7% error) for molecular crystals
- This success occurs despite measurable many-body effects in small clusters
How can pairwise models work so well for crystals when we just showed many-body effects are important? The answer lies in error cancellation, and the nature of crystals vs. trimers:
- Many-body effects may cancel - e.g. while the net polarisation of a molecule from 3 molecules might be very different than 2, it may be that for a full crystal these largely cancel... so:
- Collective effects average out - individual cooperative/anti-cooperative terms partially cancel
This is why understanding fundamentals matters - it helps you recognize when approximations will work and when they'll fail. More importantly, testing and benchmarking accuracy is fundamental to understanding the success and potential failures of different models!
The CE-1p Model for Crystals
The CE-1p model extends naturally from dimers to crystals by summing pairwise interactions. It captures the essential physics while remaining computationally efficient:
- Input: Crystal structure + monomer wavefunctions
- Output: Lattice energy with physical decomposition
- Speed: much much cheaper computationally than e.g. full periodic DFT
- Accuracy: MAD of 3.3 kJ/mol error for molecular crystals (X23 set)
Practical Considerations and Challenges
Convergence Issues with Ionic Crystals
The NaCl example demonstrates why simple cutoff-based summation fails for ionic systems. The long-range electrostatics lead to:
- Wild energy oscillations with cutoff radius
- Dependence on summation order
- Need for Ewald summation or similar techniques
Polymorphism: When Small Differences Matter
Many pharmaceutical compounds have multiple crystal forms (polymorphs) with energy differences < 5 kJ/mol. Since RT ≈ 2.5 kJ/mol at room temperature, thermal effects can reverse stability rankings. This is why:
- Temperature-dependent studies are crucial, and vibrational enthalpy contributions are important.
- Actual growth conditions (nucleation), kinetic factors often control which form crystallizes
- Computational predictions need ~1 kJ/mol accuracy
Visualizing Crystal Energetics
Tools like CrystalExplorer can generate "energy frameworks" - 3D visualizations where tube thickness represents interaction strength. This reveals:
- Hydrogen bonding networks
- π-π stacking columns
- Mechanical anisotropy origins
Key Takeaways
✓ Energy units matter - always specify per molecule, per formula unit, or per atom
✓ Pairwise models can work surprisingly well - CE-1p achieves ~10% accuracy by smart parameterization
✓ Convergence differs by system - molecular crystals converge smoothly, ionic crystals oscillate wildly
✓ Small energy differences have big consequences - polymorphs often differ by < RT
✓ Connect to experiment through proper thermodynamics - don't forget the 2RT term!
Ready to see how crystals actually grow? Let's explore nucleation and growth mechanisms!