3. Crystal Growth: Putting It All Together
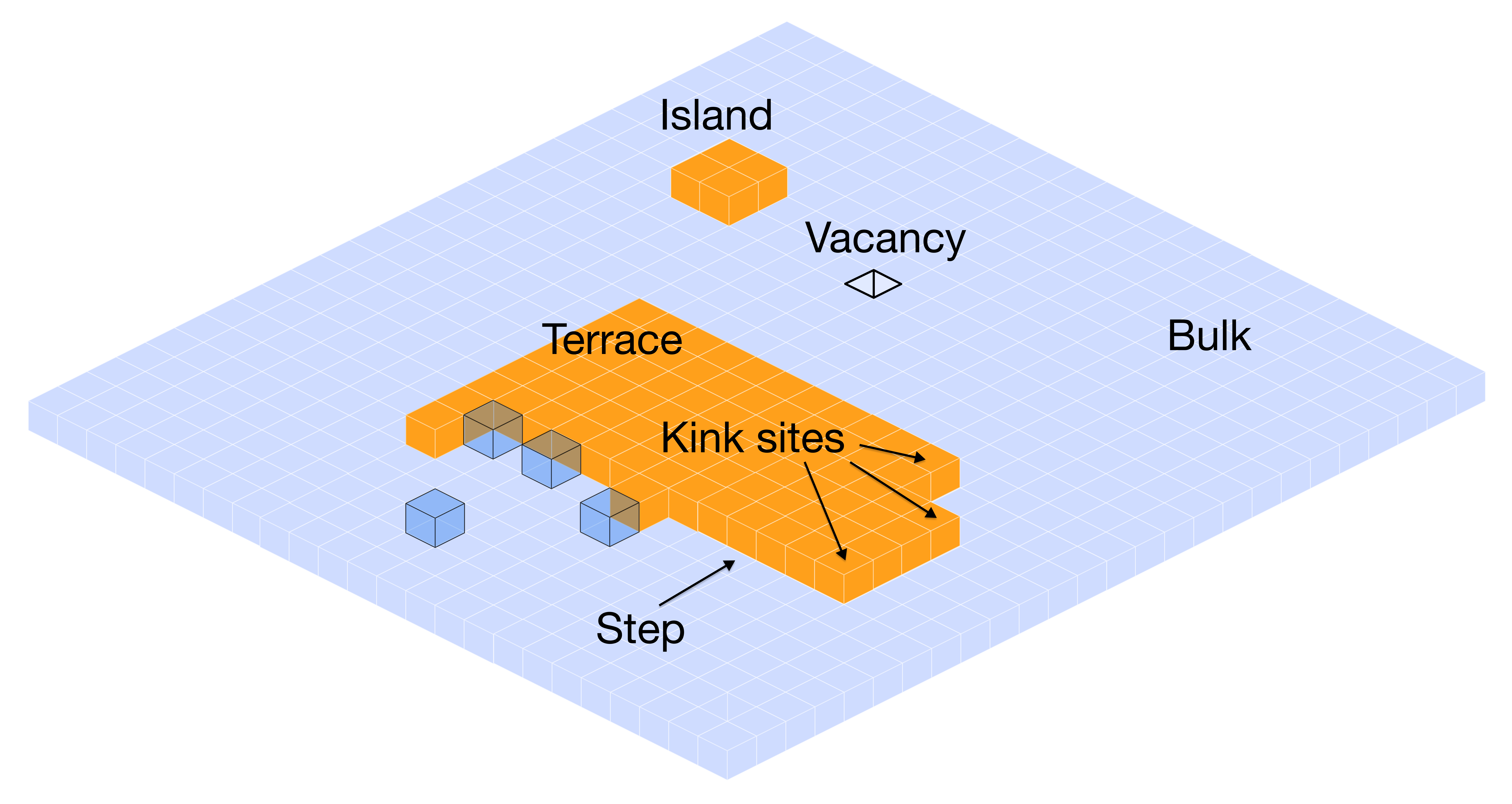
How do molecules assemble into crystals? This final section combines our understanding of intermolecular interactions and crystal energetics to predict how crystals grow, what shapes they form, and how solvents affect the process.
From Molecules to Crystals: The Growth Process
Crystal growth involves several key steps, each governed by the intermolecular interactions we've studied:
- Nucleation: Small molecular clusters form in solution - governed by dimer and trimer energies
- Attachment: New molecules join crystal surfaces - attachment energy determines growth rate
- Surface integration: Molecules find stable positions - surface energy controls final morphology
- Solvent competition: Solvent molecules compete with crystal attachment - affects both growth rate and shape
The key insight is that slow-growing faces dominate the final crystal morphology because fast-growing faces quickly disappear. This is why attachment energies (how strongly molecules stick to different faces) directly determine crystal shape.
Computational Exercises
We'll use paracetamol (acetaminophen) to demonstrate how our understanding of intermolecular interactions predicts crystal growth behavior.
Exercise 1: Solvation Free Energy Calculations
Understanding how molecules interact with solvents is crucial for predicting solubility and crystal growth behavior. We'll calculate the solvation free energy of paracetamol in water using the SMD (Solvation Model based on Density) approach.
SMD Solvation Energy Calculation
Calculate the solvation free energy of paracetamol in water to understand solvent stabilization effects and their impact on drug solubility.
Step 1: Navigate to the SMD directory
Instructions
Navigate to the directory containing the prepared input files for gas-phase and solvated calculations.
Commands
cd awscc_workshop_2025/03_crystal_growth/SMD/
ls -la
head -4 gas.inp smd.inpExpected Output
gas.inp smd.inp paracetamol.xyz run_smd.sh
==> gas.inp <==
! wB97M-V DEF2-SVP
* XYZFILE 0 1 paracetamol.xyz
==> smd.inp <==
! wB97M-V DEF2-SVP SMD(WATER)
* XYZFILE 0 1 paracetamol.xyzNotes
The only difference is SMD(WATER) in the solvated calculation input file. Note that the smd input file will be modified/regenerated by the script when run.
Exercise 2: Crystal Growth and Morphology Prediction
Crystal morphology - the external shape of crystals - determines properties like dissolution rate, flowability, and bioavailability. We'll predict paracetamol crystal morphology using surface energy calculations and examine how solvents affect crystal growth.
Crystal Growth Morphology Prediction
Calculate surface energies for different crystal faces of paracetamol to predict crystal morphology and understand solvent effects on crystal growth.
Step 1: Navigate to crystal growth directory
Instructions
Navigate to the CG directory containing the crystal structure and examine the setup for morphology calculations.
Commands
cd ../CG/
ls -la
head -20 paracetamol.cifExpected Output
paracetamol.cif paracetamol_dimers/ run_paracetamol_cg.sh
data_paracetamol
_chemical_name_common Acetaminophen
_chemical_formula_moiety 'C8 H9 N1 O2'
_chemical_name_systematic N-(4-Hydroxyphenyl)acetamide
_chemical_properties_biological 'antiinflammatory agent'
_diffrn_ambient_temperature 295
_exptl_crystal_density_diffrn 1.293
#These two values have been output from a single CSD field.
_refine_ls_R_factor_gt 0.072
_refine_ls_wR_factor_gt 0.072
_diffrn_radiation_probe x-ray
_symmetry_cell_setting monoclinic
_symmetry_space_group_name_H-M 'P 21/a'
_symmetry_Int_Tables_number 14
_space_group_name_Hall '-P 2yab'
loop_
_symmetry_equiv_pos_site_id
_symmetry_equiv_pos_as_xyz
1 x,y,z
2 1/2-x,1/2+y,-zNotes
Paracetamol Form I has monoclinic symmetry (P21/a) with different a, b, and c dimensions.
Understanding the Results
The crystal growth calculations demonstrate how molecular-level interactions determine macroscopic crystal properties. The OCC calculation provides both vacuum and solvated surface energies, showing how solvent dramatically affects which crystal faces dominate the final morphology.
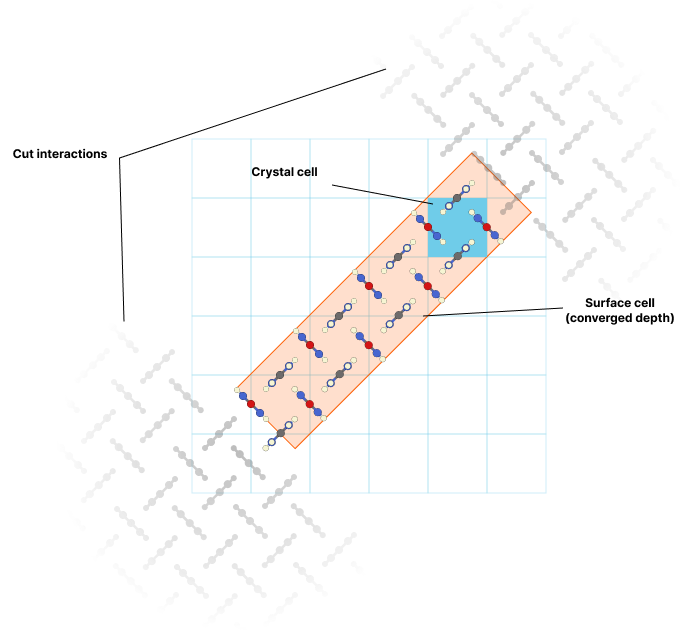
Notice in the output that each surface has a "cut" parameter (e.g., cut = 0.250 for the (1 0 0) face). This represents where along the unit cell the surface is cleaved, as different cut positions expose different molecular groups and have vastly different energies. Finding the optimal cut position for each face is computationally intensive but crucial for accurate morphology prediction.
The key insight is that faces with lower surface energies grow more slowly and therefore become the dominant faces in the final crystal. Water affects different faces differently - some are stabilized by 60% while others by only 18% - which explains why crystals grown from different solvents have different shapes.
Computational Advantages
What we just accomplished in minutes would be extraordinarily challenging with traditional periodic DFT methods. Consider what periodic DFT would require:
- Surface slab construction: Building periodic slabs with sufficient vacuum space and thickness to prevent artifacts
- Solvation modeling: Explicit water molecules or implicit solvation models that work poorly with periodic boundaries
- Convergence challenges: Solvated surfaces often require hundreds of optimization steps
- Computational cost: Each surface would require hours to days of calculation time
- Multiple cut positions: Testing different surface terminations multiplies the computational burden
In contrast, the OCC approach leverages accurate gas-phase molecular wavefunctions and well-validated solvation models to predict surface energies efficiently. The method handles both vacuum and solvated surfaces in a single calculation, automatically finding optimal cut positions and providing results in minutes rather than weeks.
Applications and Real-World Impact
Understanding and predicting crystal morphology has profound implications for pharmaceutical development. The shape of drug crystals directly affects how quickly tablets dissolve in the body, how well powders flow during manufacturing, and even how stable the drug remains on the shelf. By using computational methods like those demonstrated here, pharmaceutical companies can predict these properties before synthesizing a single crystal.
The power of this approach lies in its ability to screen many different crystallization conditions rapidly. Rather than spending months growing crystals from dozens of solvents, researchers can computationally predict which conditions will produce crystals with desired properties. The surface energy calculations reveal not just what shape crystals will form, but why - providing the physical understanding needed to rationally design crystallization processes.
This same methodology extends beyond pharmaceuticals to any crystalline material where morphology matters: semiconductors for electronics, explosives for safety applications, and specialty chemicals where crystal shape affects performance. The computational approach provides both practical predictions and fundamental understanding of the crystal growth process.
Key Takeaways
✓ Crystal faces with lower surface energies grow slowly and dominate the final morphology
✓ Solvents dramatically alter surface energies, changing crystal shape
✓ OCC efficiently predicts both vacuum and solvated morphologies in minutes
✓ The PLY output files provide 3D visualizations of predicted crystal shapes
For detailed information about the theoretical foundation and computational methodology behind crystal growth calculations, see the Crystal Growth Theory appendix.